
国家药监局决定不同新药监测期的依据是A.临床前研究的一般资料B.现有的注册资料C.现有的安全性研究资料,境内外研究状况D.境内外安全性研究状况E.现有的安全性研究资料
题目
国家药监局决定不同新药监测期的依据是
A.临床前研究的一般资料
B.现有的注册资料
C.现有的安全性研究资料,境内外研究状况
D.境内外安全性研究状况
E.现有的安全性研究资料
相似考题
更多“国家药监局决定不同新药监测期的依据是A.临床前研究的一般资料B.现有的注册资料C.现有的安全性研 ”相关问题
-
第1题:
下列哪一项不是申办者在临床试验前必须准备和提供的?()
A.试验用药品
B.受试者的个人资料
C.该药已有的临床资料
D.该药的临床前研究资料
答案B -
第2题:
化学药品注册分类I类至V类新药审批的一般程序是( )。
A.提供符合规定的综述资料、药学研究资料药理毒理研究资料、临床研究资料,向SFDA申请临床试验批文
B.申办者选择或由SFDA指定具有资质的国家药品研究基地为临床研究单位
C.取得SFDA批文后进行临床研究(I、Ⅱ、Ⅲ期)
D.提供临床研究资料及临床前研究资料,经审评合格取得试生产批文,试生产期间进行药品生产质量观察及Ⅳ期临床试验,并在试生产满前3个月,提供有关资料申请转正
E.审批合格后转为正式生产
正确答案:ABCDE
-
第3题:
新药的监测期根据现有的安全性研究资料和境内外研究状况确定,自新药批准生产之日起计算,最长不得超过
A、1年
B、2年
C、3年
D、5年
E、10年
参考答案:D
-
第4题:
新药申请注册的程序除新药生产申请外,还有的是
A、新药临床试验申请
B、资料申报
C、技术审评
D、初查和现场核查
E、省级药品监督管理部门的形式审查
参考答案:A
-
第5题:
新药临床研究的审批过程中申请人的职责是
A.完成临床前研究
B.填写《药品注册申请表》
C.对抽取的样品进行检验
D.向所在地省级药监局如实报送有关资料
E.报送药物实样
正确答案:ABDE
-
第6题:
容许新药研究者使用微剂量(一般不大于100μg)、在少量受试者(6人左右)进行药物试验。目的是评价药物的安全性和药动学特征,降低新药研究风险,提高新药研究开发命中率的是A.毒理学研究
B.Ⅰ期临床试验
C.Ⅲ期临床试验
D.Ⅳ期临床试验
E.0期临床试验答案:E解析:本题考查的是新药的研究与开发。0期临床试验属于探索性实验,目的是保证Ⅰ期临床试验的安全性。其特点是不以评价疗效为目的,主要评价药物的安全性和药动学特点。 -
第7题:
为人体安全性评价,一般选20~30例健康成年人志愿者,为制定临床给药方案提供依据,称为A.Ⅰ期临床研究
B.Ⅱ期临床研究
C.Ⅲ期临床研究
D.Ⅳ期临床研究
E.0期临床研究答案:A解析:新药临床试验一般分为4期:Ⅰ期临床试验为人体安全性评价,一般选20~30例健康成年人志愿者,为制定临床给药方案提供依据;Ⅱ期临床试验为初步药效学评价试验,大于100例实验,推荐临床给药剂量。 -
第8题:
利用已有的资料或通过特殊调查得到的资料,按不同的地区、时间、人群特征分组,把疾病和健康状况的分布情况真实地展现出来的研究方法是( )。A.分析性研究
B.描述性研究
C.临床试验研究
D.现场试验研究
E.社区试验研究答案:B解析:描述性研究是指利用已有的资料或通过特殊调查得到的资料按不同的地区、时间、人群特征分组,把疾病或健康状态的分布情况真实地展现出来。描述性研究是流行病学研究的基本步骤,常用的描述性研究包括现况研究和筛检。 -
第9题:
关于药物的临床试验叙述正确的是A.须经国家药监局批准,必须执行《药物临床试验质量管理规范》
B.临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期
C.新药在批准上市前,应当进行Ⅰ、Ⅱ、Ⅲ期临床试验
D.Ⅲ期为治疗作用确证阶段答案:A,B,C,D解析:临床试验是决定候选药物能否成为新药上市销售的关键阶段,这一阶段必须获得国家药物监督管理部门的批准在具有临床药物临床试验资格的机构中实施,并严格遵守《药物临床试验质量管理规定》的规定。临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。新药在批准上市前,申请新药注册应当完成Ⅰ、Ⅱ、Ⅲ期临床试验。Ⅲ期为治疗作用确证阶段。故选ABCD。 -
第10题:
描述性研究是用已有的资料或专项调查资料按不同地区、不同时间及不同人群特征分组,以发病率、死亡率、构成比等指标把疾病和健康状况展示出来,为病因研究提供线索,为分析性研或实验研究提出假设,为制定治疗、护理、预防措施提供科学依据。
正确答案:正确 -
第11题:
新药的申报与审批,以下说法不符合2005年《药品注册管理办法》的是()
- A、突发事件应急所必需的药品国家食品药品监督管理局可以实行快速审批
- B、申请人应依次申请《药物临床试验批件》、《药品注册批件》、新药证书与药品批准文号
- C、新药药品说明书由申请人提出,国家食品药品监督管理局对药品说明书予以核准,申请人对药品说明书正确性与准确性负责
- D、新药的监测期根据现有的安全性研究资料和境内外研究状况确定,自新药批准生产之日起计算为5年
正确答案:D -
第12题:
填空题新药研究开发过程分为()、临床前研究、临床研究、注册上市和上市后药物监测四个阶段。正确答案: 研究方针确定和发现有效的药物解析: 暂无解析 -
第13题:
新药临床研究的审批过程中属于省局药监局的职责有
A.对抽取的样品进行检查
B.对申报资料进行形式审查
C.向确定的药品检验所发出注册检验通知
D.填写《药品注册申请表》
E.将审查意见、考察报告及申报资料报送国家药监局
正确答案:BCE
-
第14题:
新药进行临床试验必须提供()
A.系统药理研究数据
B.新药作用谱
C.临床前研究资料
D.LD50
E.急慢性毒理研究数据
正确答案:C
-
第15题:
药物安全性评价应包括
A.新药临床评价和药物上市后再评价
B.药物临床评价
C.新药的临床前研究
D.临床评价和实验室评价
E.临床前研究和上市后药品的实验室评价
正确答案:D
-
第16题:
化学药品注册分类I至V类新药审批的一般程序是
A.提供符合规定的综述资料、药学研究资料、药理毒理研究资料、临床研究资料,向SFDA申请临床试验批文
B.申办者选择或由SFDA指定具有资质的国家药品研究基地为临床研究单位
C.取得SFDA批文后进行临床研究(Ⅰ、Ⅱ、Ⅲ期)
D.提供临床研究资料及临床前研究资料,经审评合格取得试生产批文试生产期间进行药品生产质量观察及Ⅳ期临床试验,并在试生产满前3个月,提供有关资料申请转正
E.审批合格后转为正式生产
正确答案:ABCDE
-
第17题:
新药进行临床试验必须提供A.系统药理研究数据
B.急慢性毒性观察结果
C.新药作用谱
D.LD.50
E.临床前研究资料答案:E解析:新药进行临床试验必须提供临床前研究资料。 -
第18题:
为扩大的多中心临床试验,完成例数大于300例,为新药注册申请提供依据,称为A.Ⅰ期临床研究
B.Ⅱ期临床研究
C.Ⅲ期临床研究
D.Ⅳ期临床研究
E.0期临床研究答案:C解析:Ⅲ期临床试验为扩大的多中心临床试验,大于300例,为新药注册申请提供依据;Ⅳ期临床试验为批准上市后的监测,也称售后调研,对最终确定新药的临床价值有重要意义;0期实验,先于Ⅰ期临床试验,评价试药的安全性和药动学性质,不以药效评价为目的,6人左右,剂量不大于100μg。 -
第19题:
对已有的资料或者疾病监测记录做分析或总结称为A.横断面研究
B.纵向研究
C.常规资料分析
D.病例对照研究
E.群组研究答案:C解析: -
第20题:
新药进行临床试验必须提供A.系统药理研究数据
B.急慢性毒性观察结果
C.新药作用谱
D.LD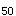
E.临床前研究资料答案:E解析: -
第21题:
()是决定候选药物能否成为新药上市销售的关键阶段。A.Ⅳ期临床试验
B.Ⅰ期临床试验
C.药理毒理研究
D.药品再注册答案:C解析:药物临床试验是指要以药品上市注册为目的,为确定药物安全性与有效性在人体开展的药物研究。药物临床试验是决定候选药物能否成为新药上市销售的关键阶段。故选C。 -
第22题:
新药开发的过程包括:()
- A、新药临床前研究和新药临床研究
- B、新药临床前研究
- C、新药临床研究
- D、新药临床试验Ⅰ期
正确答案:A -
第23题:
新药研究开发过程分为()、临床前研究、临床研究、注册上市和上市后药物监测四个阶段。
正确答案:研究方针确定和发现有效的药物